Comparaisons multiples et reproductibilité#
Tester une seule hypothèse à 5% de risque d’erreur, c’est raisonnable. Mais que se passe-t-il quand on effectue 20, 100, ou 10 000 tests simultanément — comme en génomique, en neuro-imagerie, ou lors d’analyses exploratoires de données ? La probabilité d’obtenir au moins un faux positif explose. Ce chapitre présente les méthodes de correction pour comparaisons multiples, illustre le problème du p-hacking, et aborde la crise de la reproductibilité.
Le problème des comparaisons multiples#
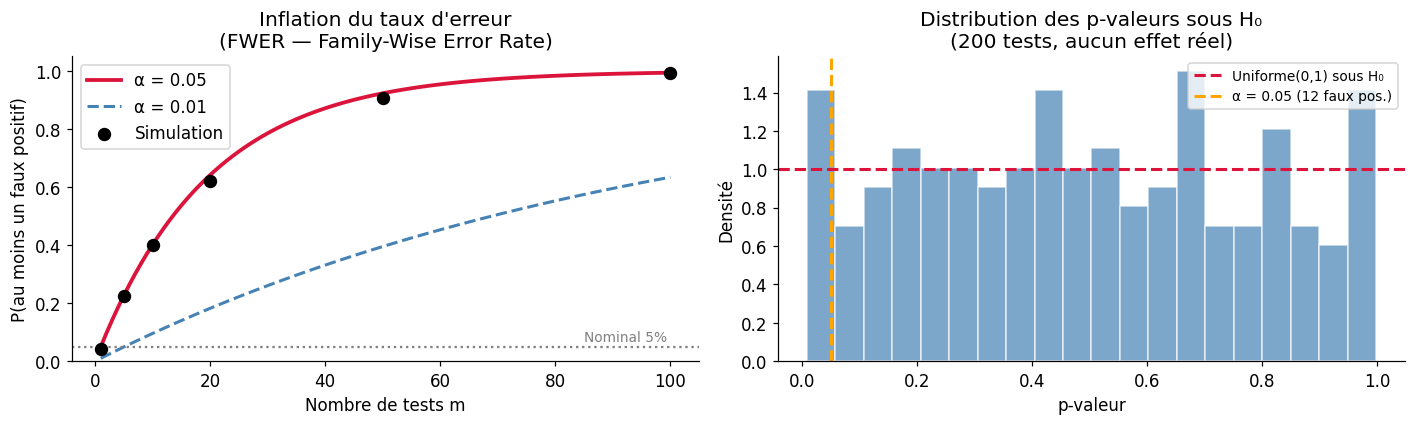
Inflation du taux d’erreur de type I#
Si on effectue \(m\) tests indépendants, chacun au seuil \(\alpha\), la probabilité d’obtenir au moins un faux positif est :
Pour \(\alpha = 0{,}05\) et \(m = 20\) tests : \(1 - 0{,}95^{20} \approx 64\%\).
# Démonstration par simulation : taux de faux positifs réels
def simulation_faux_positifs(m_tests, alpha=0.05, n_simul=500, n_obs=30):
"""Simule m_tests sous H0 vraie et compte les faux positifs (vectorisé)."""
# Génère toutes les données en une seule fois : (n_simul, m_tests, 2, n_obs)
data = rng.normal(0, 1, (n_simul, m_tests, 2, n_obs))
g1, g2 = data[:, :, 0, :], data[:, :, 1, :]
# Welch t-test vectorisé
m1, m2 = g1.mean(axis=2), g2.mean(axis=2)
s1, s2 = g1.var(axis=2, ddof=1), g2.var(axis=2, ddof=1)
se = np.sqrt(s1 / n_obs + s2 / n_obs)
t = (m1 - m2) / se
df = (s1 / n_obs + s2 / n_obs)**2 / (
(s1 / n_obs)**2 / (n_obs - 1) + (s2 / n_obs)**2 / (n_obs - 1)
)
p_valeurs = 2 * stats.t.sf(np.abs(t), df=df) # (n_simul, m_tests)
return (p_valeurs < alpha).sum(axis=1) # nb faux positifs par simulation
m_values = [1, 5, 10, 20, 50, 100]
taux_fwer = []
for m in m_values:
fp = simulation_faux_positifs(m, n_simul=500)
taux_fwer.append((fp >= 1).mean())
# Valeurs théoriques
taux_theorique = [1 - (1 - 0.05)**m for m in m_values]
print("Nombre de tests | Taux FWER simulé | Taux FWER théorique")
print("-" * 55)
for m, sim, theo in zip(m_values, taux_fwer, taux_theorique):
print(f"{m:>15} | {sim:>16.3f} | {theo:>19.3f}")
Nombre de tests | Taux FWER simulé | Taux FWER théorique
-------------------------------------------------------
1 | 0.042 | 0.050
5 | 0.224 | 0.226
10 | 0.400 | 0.401
20 | 0.622 | 0.642
50 | 0.908 | 0.923
100 | 0.994 | 0.994
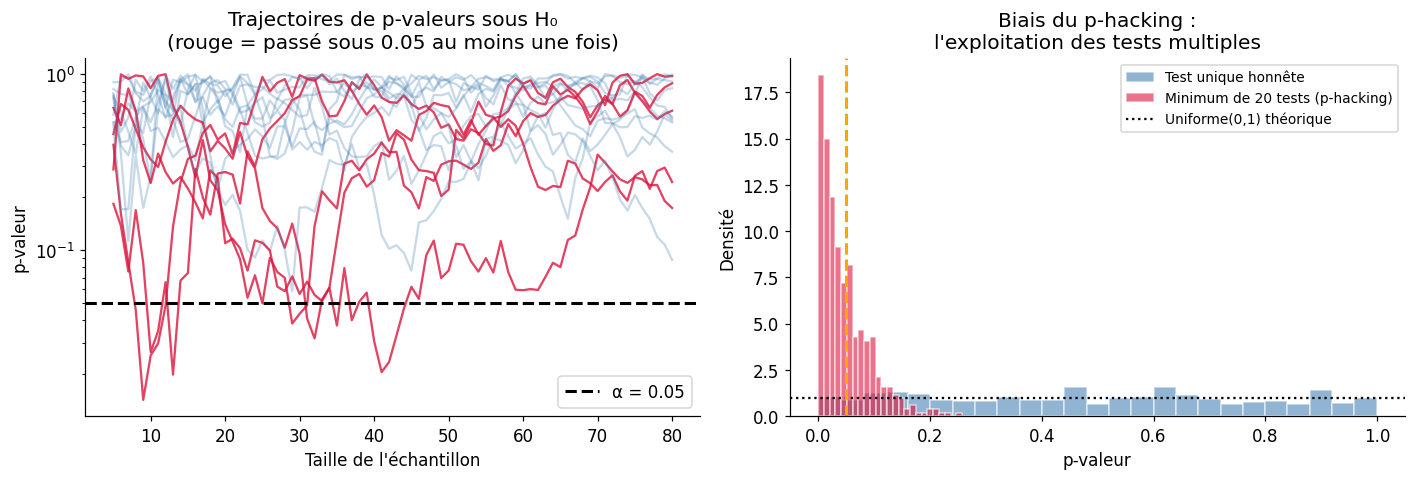
Le p-hacking : démonstration#
Le p-hacking consiste à chercher des analyses ou des sous-groupes jusqu’à obtenir \(p < 0{,}05\), puis à ne rapporter que ce résultat. C’est une forme de biais de publication et une menace majeure pour la reproductibilité.
Avec test répété jusqu'à n=100 (H₀ vraie) :
Taux de 'découvertes' : 31.6%
(devrait être 5% pour un test unique honnête)
Correction de Bonferroni#
La correction la plus simple : diviser \(\alpha\) par le nombre de tests \(m\).
La correction de Bonferroni contrôle le FWER (Family-Wise Error Rate) — la probabilité d’au moins un faux positif. Elle est valable même si les tests sont corrélés (contrôle conservateur).
Conservatisme de Bonferroni
Bonferroni est très conservateur lorsque \(m\) est grand et que les tests sont corrélés positivement. Elle peut manquer des vrais positifs (faible puissance). Pour des analyses exploratoires ou en génomique, préférer les méthodes FDR.
Holm-Bonferroni : plus puissant que Bonferroni#
La procédure de Holm contrôle aussi le FWER mais est uniformément plus puissante que Bonferroni :
Trier les p-valeurs : \(p_{(1)} \leq p_{(2)} \leq \cdots \leq p_{(m)}\)
Pour \(i = 1, 2, \ldots, m\) : rejeter \(H_{(i)}\) si \(p_{(j)} \leq \alpha / (m - j + 1)\) pour tout \(j \leq i\)
Dès qu’un test n’est pas rejeté, arrêter
# Exemple : 10 tests, dont 3 avec vrais effets
rng_ex = np.random.default_rng(7)
# Simuler 10 p-valeurs : 7 sous H0, 3 avec vrais effets
n_tests_tot = 10
n_vrais_effets = 3
p_vals_sim = []
vrai_effet = []
for i in range(n_tests_tot):
if i < n_vrais_effets:
# Vrai effet : d = 0.7
g1 = rng_ex.normal(0, 1, 50)
g2 = rng_ex.normal(0.7, 1, 50)
vrai_effet.append(True)
else:
# Sous H0
g1 = rng_ex.normal(0, 1, 50)
g2 = rng_ex.normal(0, 1, 50)
vrai_effet.append(False)
_, p = stats.ttest_ind(g1, g2)
p_vals_sim.append(p)
df_tests = pd.DataFrame({
'Test': [f'H_{i+1}' for i in range(n_tests_tot)],
'p_brute': p_vals_sim,
'vrai_effet': vrai_effet
})
# Appliquer les corrections
for method, col_name in [('bonferroni', 'p_bonf'), ('holm', 'p_holm'),
('fdr_bh', 'p_fdr_bh'), ('fdr_by', 'p_fdr_by')]:
reject, pvals_corr, _, _ = multipletests(p_vals_sim, alpha=0.05, method=method)
df_tests[col_name] = pvals_corr
df_tests[f'rej_{method}'] = reject
print("Comparaison des méthodes de correction :")
print(df_tests[['Test', 'p_brute', 'p_bonf', 'p_holm', 'p_fdr_bh', 'vrai_effet']].to_string(index=False))
print()
print("Rejets au seuil 0.05 :")
for method, col in [('Sans correction', 'p_brute'), ('Bonferroni', 'p_bonf'),
('Holm', 'p_holm'), ('FDR Benjamini-Hochberg', 'p_fdr_bh')]:
n_rejets = (df_tests[col] < 0.05).sum()
n_vrais = sum(r and e for r, e in zip(df_tests[col] < 0.05, df_tests['vrai_effet']))
n_faux = n_rejets - n_vrais
print(f" {method:<30} : {n_rejets} rejets ({n_vrais} vrais positifs, {n_faux} faux positifs)")
Comparaison des méthodes de correction :
Test p_brute p_bonf p_holm p_fdr_bh vrai_effet
H_1 5.459427e-07 0.000005 0.000005 0.000005 True
H_2 2.775236e-05 0.000278 0.000222 0.000093 True
H_3 8.272765e-06 0.000083 0.000074 0.000041 True
H_4 2.968676e-01 1.000000 1.000000 0.424097 False
H_5 2.038460e-01 1.000000 1.000000 0.339743 False
H_6 3.809623e-02 0.380962 0.266674 0.095241 False
H_7 7.886507e-01 1.000000 1.000000 0.876279 False
H_8 3.709269e-01 1.000000 1.000000 0.463659 False
H_9 7.019461e-02 0.701946 0.421168 0.140389 False
H_10 8.833582e-01 1.000000 1.000000 0.883358 False
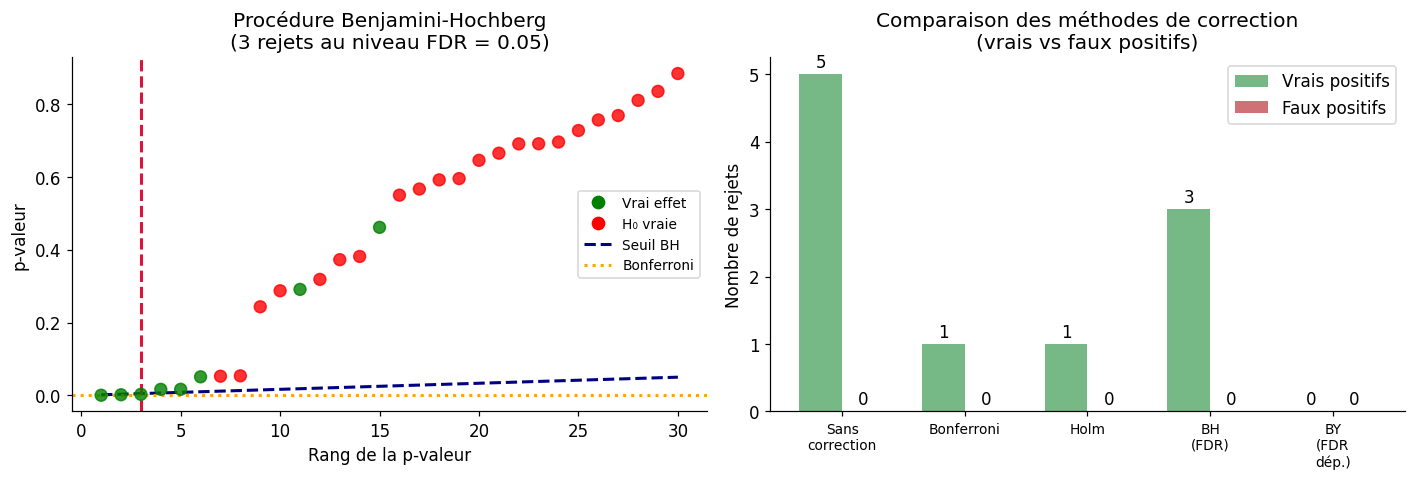
Rejets au seuil 0.05 :
Sans correction : 4 rejets (3 vrais positifs, 1 faux positifs)
Bonferroni : 3 rejets (3 vrais positifs, 0 faux positifs)
Holm : 3 rejets (3 vrais positifs, 0 faux positifs)
FDR Benjamini-Hochberg : 3 rejets (3 vrais positifs, 0 faux positifs)
FDR — Benjamini-Hochberg#
Le Taux de Faux Découvertes (False Discovery Rate, FDR) est plus adapté aux analyses à grande échelle. Plutôt que d’interdire tout faux positif, on contrôle la proportion de fausses découvertes parmi les rejets.
La procédure de Benjamini-Hochberg :
Trier les p-valeurs : \(p_{(1)} \leq p_{(2)} \leq \cdots \leq p_{(m)}\)
Trouver le plus grand \(k\) tel que \(p_{(k)} \leq \frac{k}{m} \cdot q^*\)
Rejeter \(H_{(1)}, \ldots, H_{(k)}\)
où \(q^*\) est le niveau FDR cible (souvent 0,05 ou 0,10).
Volcano plot : visualiser les résultats à grande échelle#
Le volcano plot est la visualisation standard en bioinformatique et en pharmacologie pour représenter simultanément la taille d’effet et la significativité statistique.
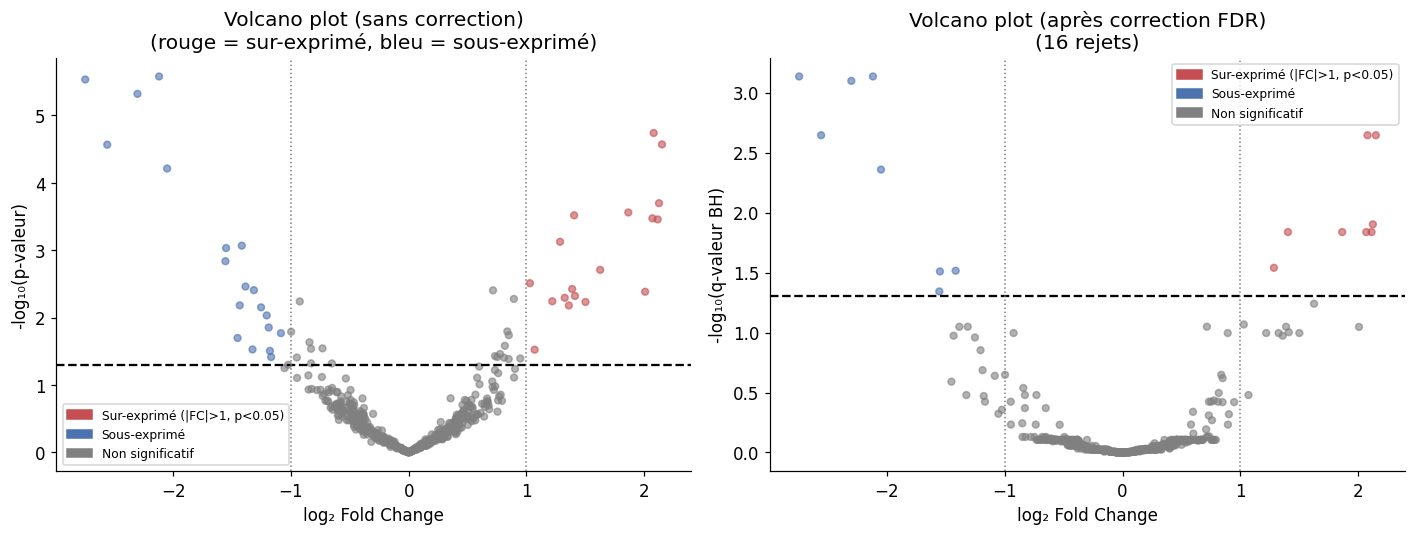
Sans correction : 37 gènes significatifs (|FC|>1)
Après FDR BH : 16 gènes significatifs (|FC|>1)
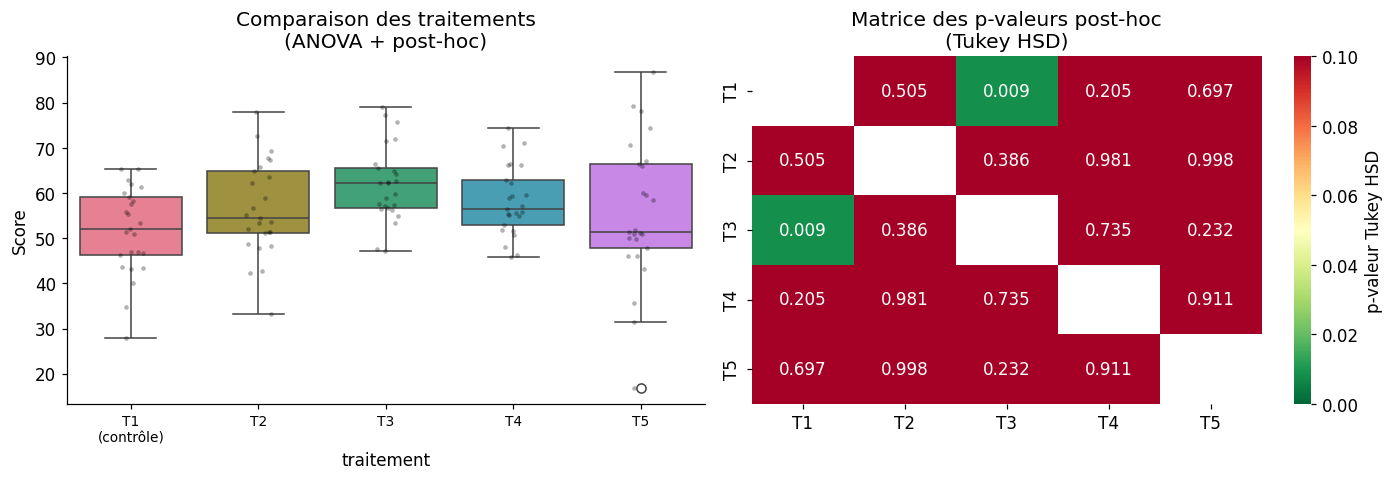
Tests post-hoc ANOVA#
Après une ANOVA significative, on identifie les paires différentes avec des tests post-hoc spécialisés, qui contrôlent les comparaisons multiples par construction.
# Exemple : 5 traitements médicaux, mesure sur 25 patients chacun
rng_ph = np.random.default_rng(123)
n_pat = 25
traitements = {
'T1 (contrôle)': rng_ph.normal(50, 10, n_pat),
'T2': rng_ph.normal(55, 10, n_pat),
'T3': rng_ph.normal(62, 10, n_pat),
'T4': rng_ph.normal(58, 10, n_pat),
'T5': rng_ph.normal(55, 14, n_pat), # variance plus grande
}
df_ph = pd.DataFrame({
'score': np.concatenate(list(traitements.values())),
'traitement': np.repeat(list(traitements.keys()), n_pat)
})
# ANOVA globale
aov_ph = pg.anova(data=df_ph, dv='score', between='traitement')
print("ANOVA globale :")
print(aov_ph[['Source', 'F', 'p_unc', 'np2']].to_string(index=False))
print()
# Tukey HSD
print("Tukey HSD :")
tukey_ph = pg.pairwise_tukey(data=df_ph, dv='score', between='traitement')
print(tukey_ph[['A', 'B', 'diff', 'se', 'T', 'p_tukey']].to_string(index=False))
print()
# Games-Howell (variances inégales)
print("Games-Howell (robuste aux variances inégales) :")
gh = pg.pairwise_gameshowell(data=df_ph, dv='score', between='traitement')
print(gh[['A', 'B', 'diff', 'se', 'T', 'pval']].to_string(index=False))
ANOVA globale :
Source F p_unc np2
traitement 3.047068 0.019695 0.092204
Tukey HSD :
A B diff se T p_tukey
T1 (contrôle) T2 -4.869111 3.057277 -1.592630 0.505027
T1 (contrôle) T3 -10.333994 3.057277 -3.380130 0.008524
T1 (contrôle) T4 -6.578402 3.057277 -2.151720 0.205482
T1 (contrôle) T5 -3.947462 3.057277 -1.291169 0.697144
T2 T3 -5.464882 3.057277 -1.787500 0.385633
T2 T4 -1.709291 3.057277 -0.559089 0.980605
T2 T5 0.921650 3.057277 0.301461 0.998179
T3 T4 3.755591 3.057277 1.228411 0.734850
T3 T5 6.386532 3.057277 2.088961 0.231633
T4 T5 2.630941 3.057277 0.860550 0.910629
Games-Howell (robuste aux variances inégales) :
A B diff se T pval
T1 (contrôle) T2 -4.869111 2.844908 -1.711518 0.436823
T1 (contrôle) T3 -10.333994 2.546409 -4.058262 0.001663
T1 (contrôle) T4 -6.578402 2.467859 -2.665631 0.075131
T1 (contrôle) T5 -3.947462 3.716810 -1.062056 0.824545
T2 T3 -5.464882 2.677213 -2.041258 0.263319
T2 T4 -1.709291 2.602614 -0.656759 0.964451
T2 T5 0.921650 3.807617 0.242054 0.999209
T3 T4 3.755591 2.272508 1.652619 0.472309
T3 T5 6.386532 3.590075 1.778941 0.400982
T4 T5 2.630941 3.534794 0.744298 0.944396
Reproductibilité et crise de la réplication#
La crise de la réplication#
En 2015, le Reproducibility Project a tenté de répliquer 100 études en psychologie : seulement ~36% ont produit des résultats significatifs similaires. Les causes identifiées :
p-hacking et researcher degrees of freedom (choix d’analyse après avoir vu les données)
Biais de publication : seuls les résultats positifs sont publiés
Taille d’échantillon insuffisante : études sous-puissantes, qui, quand elles « réussissent », surestiment les effets
Focalisation sur p < 0,05 plutôt que sur les tailles d’effet
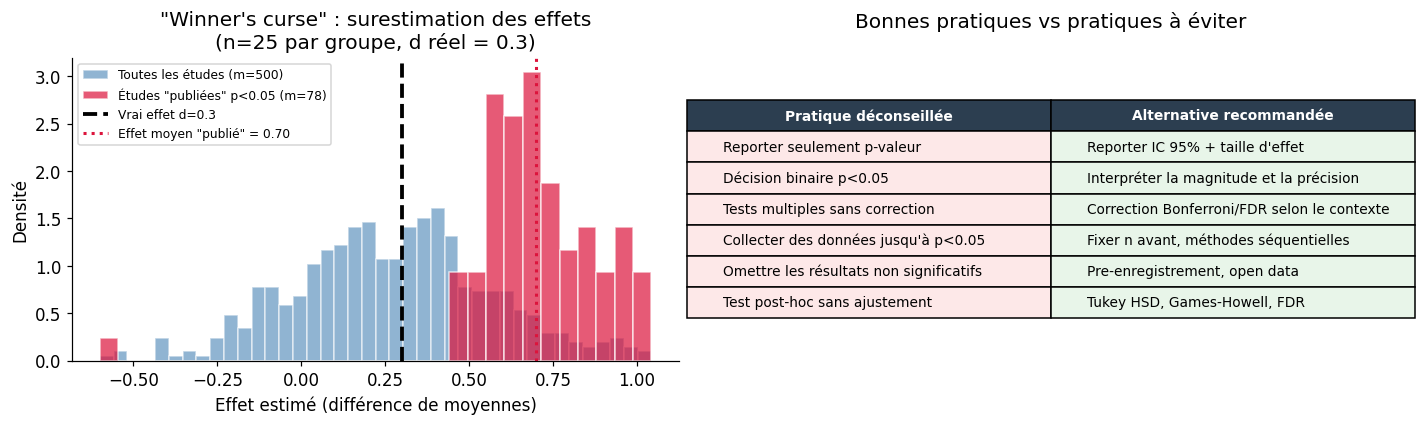
Pre-enregistrement et open science
Le pre-enregistrement consiste à déposer publiquement, avant la collecte des données, les hypothèses précises, la taille d’échantillon et la méthode d’analyse. Cela distingue les analyses confirmatives (hypothèses précises, puissance calculée) des analyses exploratoires (génération d’hypothèses, résultats préliminaires). Des plateformes comme OSF (Open Science Framework), AsPredicted, ou ClinicalTrials.gov permettent ce dépôt. Le partage des données et du code (open data, open code) est une condition supplémentaire de reproductibilité.
Résumé des méthodes de correction#
Méthode Contrôle Puissance Quand l'utiliser scipy method
Bonferroni FWER Faible Peu de tests, coût des FP élevé bonferroni
Holm-Bonferroni FWER Modérée FWER avec meilleure puissance holm
Benjamini-Hochberg FDR Bonne Analyses à grande échelle fdr_bh
Benjamini-Yekutieli FDR (dépendances) Faible Tests corrélés positivement fdr_by
Aucune — Maximale (faux positifs++) Analyse exploratoire initiale —
Usage avec statsmodels :
from statsmodels.stats.multitest import multipletests
reject, p_corr, _, _ = multipletests(p_vals, alpha=0.05, method='fdr_bh')
Comment choisir sa méthode de correction ?
Tests confirmateurs (hypothèses précises, coût d’un faux positif élevé) → Bonferroni ou Holm
Analyses exploratoires (génomique, imagerie, psychologie) → Benjamini-Hochberg (FDR)
Tests fortement corrélés (SNPs en déséquilibre de liaison, pixels voisins) → Benjamini-Yekutieli
Post-hoc ANOVA, toutes variances égales → Tukey HSD
Post-hoc ANOVA, variances inégales → Games-Howell
Post-hoc ANOVA, comparaison vs contrôle uniquement → Test de Dunnett